 During the last 10 years, we have uncovered key features of ligand recognition and structure-activity relationships of specific regulators of inflammation. The early work in the group has paved the way for our current activities which focus on the development of inhibitors with diagnostic or therapeutic potential in chronic inflammatory diseases.
During the last 10 years, we have uncovered key features of ligand recognition and structure-activity relationships of specific regulators of inflammation. The early work in the group has paved the way for our current activities which focus on the development of inhibitors with diagnostic or therapeutic potential in chronic inflammatory diseases.
The neutrophil serine proteases (NSPs) Proteinase 3 (PR3, EC 3.4.21.76) and Neutrophil Elastase (NE, EC 3.4.21.37) are therapeutic targets in a number of chronic inflammatory diseases, e.g. Chronic Obstructive Pulmonary Disease (COPD). Through 30 years, NE has been considered a drug target for COPD while it is only recently that PR3 has been indicated as a target for novel COPD therapy. WHO estimates that by 2030, COPD will be the third leading cause of death worldwide, yet there is no drug available with strong disease-modifying properties (Mathers et al., PLOS Med., 2006).
The specific roles of PR3 and NE in inflammatory diseases are only just emerging and there is a need for specific substrates that can be used in in vitro and cellular assays. Rational design of specific peptides is also a natural step towards the design of druggable low-molecular weight compounds. Yet achieving specific targeting of either of these proteases is challenging, as the mature forms of PR3 and NE share a high sequence identity (56%) and structural similarity. We have shown that differences between PR3 and NE in the nature of their S2, S1’, S2’ and S3’ substrate binding sites can be exploited to design specific substrates for PR3 (Hajjar et al., J. Med. Chem., 2006; Hajjar et al., FEBS J., 2007).
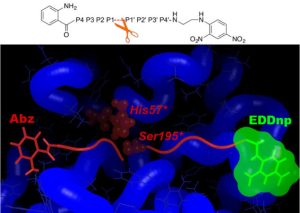
Using in silico design (MD simulations and free energy decomposition), followed by enzymatic assays, we have developed a novel FRET substrate specific for PR3. Furthermore we could show that Abz and EDDnp FRET groups significantly contribute towards substrate hydrolysis by PR3 (Narawane et al., J. Med. Chem., 2014).
Taking advantage of the knowledge and expertise built in the group we further designed ketomethylene-based peptidomimetic inhibitors for PR3 that show IC50 values in the low micromolar range. We found that the best inhibitor (Abz-VADnV[Ψ](COCH2)ADYQ-EDDnp) displayed a competitive and reversible inhibition mechanism, and it was also found to be selective for PR3 compared to NE (Budnjo et al., J. Med. Chem., 2014).
We developed an assay to perform high-throughput screening (HTS) of compound libraries on PR3 and also on NE. We then performed HTS on both proteins, which resulted in a short-list of potent inhibitors of PR3 and NE. Currently, we are driving an early drug discovery project aimed at developing novel COPD treatments.
This project is currently funded by the Norwegian Research Council through BIOTEK2021 and is part of the Center for Digital Life Norway. More information here.
Collaborators:
Véronique Witko-Sarsat (INSERM, Cochin Institute, Paris)
Bengt Erik Haug (Center for Pharmacy, University of Bergen)
Tomas M. Eagan (Department of Clinical Sciences, Haukeland University Hospital)
Anders Goksøyr (Department of Biology, University of Bergen)
Techniques:
Organic synthesis, HPLC-based and fluorescence-based assays, ELISA, docking, molecular dynamics simulations, free energy decomposition.