We are members of the screening initiatives Nor-Openscreen and EU-Openscreen as well as the X-ray crystallograpy initative NorCryst.
Please checkout the BiSS core facility to see what we can offer you in terms of fragment screening, computational chemistry and X-ray crystallography.
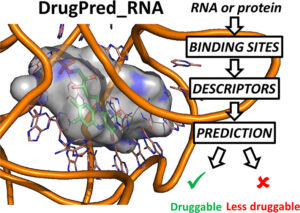 RNA-ligand interactions
RNA-ligand interactions
We are developing methods to predict the binding modes and affinities of RNA-ligand complexes and the druggabilty of RNA targets. Further, we are exploiting these methods to discover ligands for riboswitches which are novel targets for antibiotics.
RELATED NEWS PAGES:
- eHACS
- Exploration of the TPP riboswitch as a tartet for antibiotics
- Chartering chemical space for riboswitch ligands (partner project of Digital Life Norway.)
- Respond3 – Towards better computational approaches and responsible innovation strategies in early drug discovery
- Recorded lecture
SELECTED PUBLICATIONS:
- DrugPred_RNA-A tool for structure-based druggability predictions for RNA binding sites. Rekand IH, Brenk R. J Chem Inf Model. 2021. 10.1021/acs.jcim.1c00155
- Fragment-based drug discovery for RNA targets. Lundquist KP, Panchal V, Gotfredsen CH, Brenk R, Clausen MH. ChemMedChem. 2021. 10.1002/cmdc.202100324
- Ligand design for riboswitches, an emerging target class for novel antibiotics. Rekand IH, Brenk R. Future Med Chem. 2017 Sep;9(14):1649-1662. 10.4155/fmc-2017-0063
- Structure-based discovery of small molecules binding to RNA. Wehler T, Brenk R. In RNA Therapeutics; Topics in Medicinal Chemistry; Springer. 2017 pp 47–77. 10.1007/7355_2016_29
- Structure-based virtual screening for the identification of RNA-binding ligands. Daldrop P, Brenk R. Methods Mol Biol. 2014 1103, 127-39. 10.1007/978-1-62703-730-3_10
- Novel ligands for a purine riboswitch discovered by RNA-ligand docking. Daldrop P, Reyes FE, Robinson DA, Hammond CM, Lilley DM, Batey RT, Brenk R. Chem Biol. 2011 18 (3), 324-35. 10.1016/j.chembiol.2010.12.020
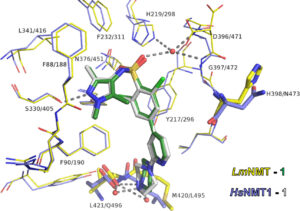 A better understanding of protein-ligand interactions
A better understanding of protein-ligand interactions
Ideally, we should be able to predict the binding affinity of a ligand based on the structure of the protein-ligand complex. However, we are still far away from reaching this goal. Therefore, we are using a combination of biophysical and computational methods to improve our understanding of protein-ligand interactions. A particular focus in this context is to better undertand the driving forces of selective inhibiton in highly conserved binding sites. In addition, we are developing methods to predict how likely it is to to find a drug-like ligand for a given binding site and which physico-chemical properties this ligand will likely have. Further, we are exploiting the vast data about protein-ligand complexes to develop scoring functions to predict the binding affinities of ligands to their targets. For this task, we are using modern machine learning methods.
RELATED NEWS PAGES:
- On the Hunt for Metalloenzyme Inhibitors
- Article published in special issue about women in medicinal chemistry
- Price for oral presentation
- Respond3 – Towards better computational approaches and responsible innovation strategies in early drug discovery
SELECTED PUBLICATIONS:
- On the hunt for metalloenzyme inhibitors: investigating the presence of metal-coordinating compounds in screening libraries and chemical spaces. Schuck B, Brenk R. Archiv der Pharmazie. 2024 e2300648. 10.1002/ardp.202300648
- How to design selective ligands for highly conserved binding sites: a case study using N-Myristoyltransferases as a model system. Kersten C, Fleischer E, Kehrein J, Borek C, Jaenicke E, Sotriffer C, Brenk R. J Med Chem. 2020 Mar 12;63(5):2095-2113. 10.1021/acs.jmedchem.9b00586
- To hit or not to hit, that is the question – Genome-wide structure-based druggability predictions for Pseudomonas aeruginosa proteins. Sarkar A, Brenk R. PLoS One. 2015 10 (9), e0137279 (2015). 10.1371/journal.pone.0137279
- DrugPred: A structure-based approach to predict protein druggability developed using an extensive nonredundant data set. Krasowski A, Muthas D, Sarkar A, Schmitt S, Brenk R. J Chem Inf Model. 2011 51 (11), 2829-42. 10.1021/ci200266d
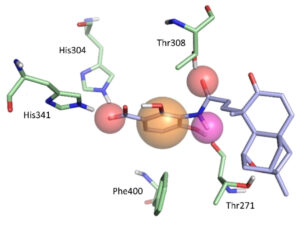 Structure-based ligand design
Structure-based ligand design
In collaboration with other labs we are working on the structure-based design of ligands for a variety of targets. A partical focus on this work is the discovery for starting points for new antibiotics.
RELATED NEWS PAGES:
- Towards the discovery of new antibiotics in the lab
- Drug Discovery get together
- Exploration of the TPP riboswitch as a tartet for antibiotics
- Structure-based exploration of new targets for antibiotics
- Webinar
SELECTED PUBLICATIONS:
- An experimental toolbox for structure-based hit discovery for P. aeruginosa FabF, a promising target for antibiotics. Espeland LO, Georgiou C, Klein R, Bhukya H, Haug BE, Underhaug J, Mainkar PS, Brenk R. ChemMedChem. 2021. 10.1002/cmdc.202100302
- Targeting the class A carbapenemase GES-5 via virtual screening. Klein R, Cendron L, Montanari M, Bellio P, Celenza G, Maso L, Tondi D, Brenk R. Biomolecules. 2020 Feb 14;10(2) pii: E304. 10.3390/biom10020304
- In silico identification and experimental validation of hits active against KPC-2 β-lactamase. Klein R, Linciano P, Celenza G, Bellio P, Papaioannou S, Blazquez J, Cendron L, Brenk R, Tondi D. PLoS One. 2018 Nov 29;13(11):e0203241. 10.1371/journal.pone.0203241
- De novo design of protein kinase inhibitors by in silico identification of hinge region-binding fragments. Urich R, Wishart G, Kiczun M, Richters A, Tidten-Luksch N, Rauh D, Sherborne B, Wyatt PG, Brenk R. ACS Chem Biol. 2013 8 (5), 1044-52. 10.1021/cb300729y
- N-myristoyltransferase inhibitors as new leads to treat sleeping sickness. Frearson JA, Brand S, McElroy SP, Cleghorn LA, Smid O, Stojanovski L, Price HP, Guther ML, Torrie LS, Robinson DA, Hallyburton I, Mpamhanga CP, Brannigan JA, Wilkinson AJ, Hodgkinson M, Hui R, Qiu W, Raimi OG, van Aalten DM, Brenk R, Gilbert IH, Read KD, Fairlamb AH, Ferguson MA, Smith DF, Wyatt PG. Nature. 2010 464 (7289), 728-32. 10.1038/nature08893