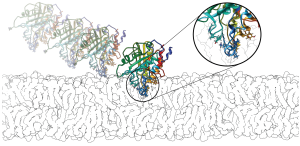
 Peripheral Membrane Proteins
Peripheral Membrane Proteins
We conduct computational and experimental studies of the membrane binding of peripheral membrane proteins. Experiments are performed in the group or conducted by collaborators. In particular, we have earned insights into the binding of serine protease 3 (PR3), the human N-acetyl transferase Naa60 and a bacterial Phospholipase C. We are also working on pattern identifications from large-scale sequence and structural data sets using bioinformatics analysis.
Peripheral membrane proteins bind temporarily to biological membranes. Unlike integral membrane proteins, the portion of the protein inserted into the lipid-bilayer is restricted to the interfacial area. In common with integral membrane proteins they share a paucity of structural data. Hence, we perform sequence and structural data analysis on peripheral proteins using bioinformatics approaches. We developed hydrophobic protrusions model (Fuglebakk and Reuter, PLoS Comp. Bio., 2018) and analysed the outcomes (Tubiana and Reuter, PLoS Comp. Bio, 2022).
We are interested in understanding the peripheral membrane binding mechanisms and the role of different amino acid types in peripheral binding. Our ongoing efforts are particularly focused on the role of aromatic amino acids and their energetic contributions in the membrane binding using free energy calculations.
Collaborators:
Experimental work for bacterial phospholipases projects are done by our external collaborators- Anne Gershenson (UMassAmherst, USA) and Mary F. Roberts (Boston College, USA).
Current work on Lipid Transfer Proteins is done in collaboration with Sergei Grudinin (NANO-D, Grenoble, France) and Anne-Claude Gavin (Unviversité de Genève, Switzerland).
TECHNIQUES:
Implicit membrane model (IMM1-GC), Continuum electrostatics, All-atom Molecular Dynamics (MD) and Coarse-Grained molecular dynamics (CGMD), Free energy calculations, Statistical analysis of protein structure databases, Surface Plasmon Resonance (SPR) Spectroscopy.
 NSPs in Chronic Inflammation
NSPs in Chronic Inflammation
During the last 10 years, we have uncovered key features of ligand recognition and structure-activity relationships of specific regulators of inflammation. The early work in the group has paved the way for our current activities which focus on the development of inhibitors with diagnostic or therapeutic potential in chronic inflammatory diseases.
The neutrophil serine proteases (NSPs) Proteinase 3 (PR3, EC 3.4.21.76) and Neutrophil Elastase (NE, EC 3.4.21.37) are therapeutic targets in a number of chronic inflammatory diseases, e.g. Chronic Obstructive Pulmonary Disease (COPD). Through 30 years, NE has been considered a drug target for COPD while it is only recently that PR3 has been indicated as a target for novel COPD therapy. WHO estimates that by 2030, COPD will be the third leading cause of death worldwide, yet there is no drug available with strong disease-modifying properties (Mathers et al., PLOS Med., 2006).
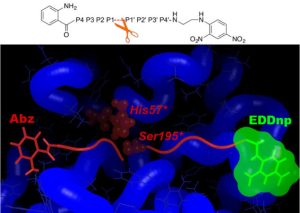
The specific roles of PR3 and NE in inflammatory diseases are only just emerging and there is a need for specific substrates that can be used in in vitro and cellular assays. Rational design of specific peptides is also a natural step towards the design of druggable low-molecular weight compounds. Yet achieving specific targeting of either of these proteases is challenging, as the mature forms of PR3 and NE share a high sequence identity (56%) and structural similarity. We have shown that differences between PR3 and NE in the nature of their S2, S1’, S2’ and S3’ substrate binding sites can be exploited to design specific substrates for PR3 (Hajjar et al., J. Med. Chem., 2006; Hajjar et al., FEBS J., 2007).
Using in silico design (MD simulations and free energy decomposition), followed by enzymatic assays, we have developed a novel FRET substrate specific for PR3. Furthermore we could show that Abz and EDDnp FRET groups significantly contribute towards substrate hydrolysis by PR3 (Narawane et al., J. Med. Chem., 2014).
Taking advantage of the knowledge and expertise built in the group we further designed ketomethylene-based peptidomimetic inhibitors for PR3 that show IC50 values in the low micromolar range. We found that the best inhibitor (Abz-VADnV[Ψ](COCH2)ADYQ-EDDnp) displayed a competitive and reversible inhibition mechanism, and it was also found to be selective for PR3 compared to NE (Budnjo et al., J. Med. Chem., 2014).
We developed an assay to perform high-throughput screening (HTS) of compound libraries on PR3 and also on NE. We then performed HTS on both proteins, which resulted in a short-list of potent inhibitors of PR3 and NE. Currently, we are driving an early drug discovery project aimed at developing novel COPD treatments.
This project is currently funded by the Norwegian Research Council through BIOTEK2021 and is part of the Center for Digital Life Norway. Check the following link for more information about this project.
COLLABORATORS:
Véronique Witko-Sarsat (INSERM, Cochin Institute, Paris)
Bengt Erik Haug (Center for Pharmacy, University of Bergen)
Tomas M. Eagan (Department of Clinical Sciences, Haukeland University Hospital)
Anders Goksøyr (Department of Biology, University of Bergen)
TECHNIQUES:
Organic synthesis, HPLC-based and fluorescence-based assays, ELISA, docking, molecular dynamics simulations, free energy decomposition, relative binding free energy calculations.


Collective motions in proteins
We develop computational methods for comparison of the proteins intrinsic dynamics, mostly through the use of elastic network models and normal mode analysis. We implemented and maintain the WEBnm@ server which allows for comparative analyses of aligned protein structures through a user-friendly interface.
Normal mode analysis (NMA) using elastic network models (ENM) is a reliable and cost-effective computational method to characterise protein flexibility and by extension, their dynamics. The conservation of the intrinsic dynamics of proteins emerges as we attempt to understand the relationship between sequence, structure and functional conservation.
Our work has been focusing on measuring the conservation of proteins flexibility, and has been supported by our activity in methods development for efficient and reliable comparison of flexibility in large structure datasets. We have early on evaluated the performances of NMA/ENM compared to principal component analysis from molecular dynamics simulations and shown that the slow dynamics was faithfully reproduced (Skjaerven, Proteins, 2011). More recently we have looked at different measures to compare protein flexibility (Fuglebakk, Bioinformatics, 2012), highlighted the impact of the choice of structural alignment in the comparison outcome (Fuglebakk, BBA General Subjects, 2015; Tiwari, Curr Op Struct Biol, 2018), and compared various ENM variants (Fuglebakk, JCTC, 2013).
Our work shows that protein flexibility characterised by its lowest frequency modes is intrinsic to protein structures and their fold (Tiwari, PLoS CB, 2016). We could show using computer-generated protein models that dynamics is highly conserved at the architecture level (Hollup, Protein Science, 2011). These models, produced by the group of Willie Taylor (Crick Institute, London) allowed us to explore a larger structure space than what is available in the Protein Data Bank.
Please check our publications on the topic for further information, in particular the three following review articles:Skjaerven, Theochem, 2009; Fuglebakk, BBA General Subjects, 2015;Tiwari, Curr Op Struct Biol, 2018.
We maintain the WEBnm@ online tool for normal mode analysis. It offers many useful analyses of normal modes, including comparative analyses of aligned proteins (Tiwari, BMC Bioinformatics, 2014). The third edition is now online in its beta version and available for testing here: WEBnma3.